Требования к производству медицинских изделий
Содержание
- 1 Производство и техобслуживание медтехники. Правовые основы
- 2 Лицензирование производства медицинской техники
- 3 Лицензирование технического обслуживания (ремонта) медицинской техники
- 4 Заявление на получение лицензии
- 5 Дополнительные документы для лицензирования
- 6 Требования, предъявляемые к соискателю лицензии
- 7 Последствия работы без лицензии



На данный момент обращение медицинских изделий, как вид предпринимательской деятельности, требует предварительного уведомления Росздравнадзора, что регламентировано Постановлением Правительства РФ №584 «Об уведомительном порядке начала осуществления отдельных видов предпринимательской деятельности», а также Федеральным законом №294 «О защите прав юридических лиц и индивидуальных предпринимателей при осуществлении государственного контроля (надзора) и муниципального контроля»: уведомление представляется после государственной регистрации и постановки на учет в налоговом органе до начала фактического выполнения работ или предоставления услуг (может быть в форме электронного документа).
За непредоставление указанного уведомления предусмотрена административная ответственность в виде наложения штрафа на должностное лицо в размере от 3 до 5 тыс.р., на юридическое лицо – от 10 до 20 тыс.р. (ч. 1 ст. 19.7.5 КоАП РФ).

Однако следует отметить, что предоставлять данное уведомление обязаны только те розничные фармацевтические организации, которые стали заниматься указанным видом деятельности позднее 18 декабря 2014 года (те, кто начал свою деятельность до указанной даты, освобождаются от уведомления).
Рассмотрим нормативно-правовое регулирование обращения медицинских изделий в разрезе основных процессов, которые происходят в розничной фармацевтической организации от момента поступления медицинского изделия до момента реализации конечному потребителю (отпуска) или уничтожения.
1. Приемка медицинских изделий.
Сама схема приемки медицинских изделий укладывается в общую схему приемки всех товаров аптечного ассортимента. Основные моменты, касающиеся порядка проведения приемочного контроля изложены в Приказе 647н «Об утверждении Правил надлежащей аптечной практики лекарственных препаратов для медицинского применения». Весь процесс начинается с проверки соблюдения условий транспортировки, соответствия поступивших медицинских изделий данным в сопроводительных документах (очень важно в данном случае проверить наличие их в информационных базах данных о приостановлении или изъятии из обращения медицинских изделий).

После проверки наличия поступивших медицинских изделий в указанных информационных базах данных и выявления положительного результата необходимо переместить медицинское изделие в зону карантинного хранения. При отрицательном результате приемочный контроль продолжается и будет состоять из следующих стадий: контроль качества, визуальный осмотр, целостность упаковки, маркировка и проверка сопроводительной документации.
Если по перечисленным параметрам медицинское изделие соответствует установленным требованиям, оно приходуется и размещается на основное место хранения; если нет – перемещается в зону карантинного хранения и либо возвращается поставщику (на условиях, прописанных в договоре) и уничтожается, либо возвращается в обращение (если поступила информация о возобновлении обращения).
Рассмотрим подробнее информационные базы данных о фальсифицированных, контрафактных и недоброкачественных медицинских изделиях.
На официальном сайте Федеральной службы по надзору в сфере здравоохранения (Росздравнадзор) есть раздел «Медицинские изделия» (подраздел «Контроль за обращением медицинских изделий», электронный сервис «Информационные письма о медицинских изделиях»).

Если на этом этапе выясняется наличие медицинского изделия в базе данных, согласно Федеральному закону №323 «Об основах охраны здоровья граждан» оно не имеет права реализовываться и в дальнейшем фальсифицированные и недоброкачественные медицинские изделия подлежат изъятию и уничтожению или вывозу, а контрафактные – изъятию и только последующему уничтожению.
Порядок уничтожения регламентирован Постановлением Правительства №1360 «Об отдельных вопросах противодействия обороту фальсифицированных, недоброкачественных и контрафактных медицинских изделий».


Один из параметров качества медицинского изделия – маркировка (именно этот параметр требует особого разъяснения, так как на данный момент нет единого нормативного документа, который содержал бы совокупную информацию о том, какая маркировка на медицинских изделиях должна быть).
Первый нормативный документ, регламентирующий маркировку – Постановление Правительства №1037 «О мерах по обеспечению наличия на ввозимых на территорию РФ непродовольственных товарах информации на русском языке», который говорит о том, что все ввозимые на территорию Российской Федерации и предназначенные для дальнейшей реализации медицинские изделия должны содержать определенный перечень данных на русском языке, а также указывает, где данная информация должна присутствовать.





Также хотелось бы обратить внимание на еще один документ – Решение Совета Евразийской экономической комиссии №30, который посвящен порядку формирования и ведения единой информационной системы в сфере обращения медицинских изделий. В п. 14 Раздела II документа присутствует информация о перечне сведений, которые должны быть указаны в данной информационной системе:
«Единый реестр содержит следующие сведения:
у) изображение маркировки медицинского изделия (в электронном виде)»

Далее рассмотрим нормативные акты, регламентирующие сопроводительные документы к медицинским изделиям.
В целом данные нормативные акты можно разделить на две группы:
– касающиеся порядка регистрации информации, которая должна присутствовать и которую мы должны предоставлять потребителю;
– касающиеся подтверждения соответствия поступающих медицинских изделий.
Согласно Постановлению Правительства №55 «Об утверждении Правил продажи отдельных видов товаров…», на медицинское изделие должна быть предоставлена информация о номере и дате регистрационного удостоверения.



Следует помнить, что регистрационные удостоверения, имеющие ограниченный срок действия и выданные до момента вступления в силу Постановления Правительства №1416 "Об утверждении правил государственной регистрации МИ», действуют до истечения их срока действия.
Регистрационные удостоверения бессрочного действия, выданные до дня вступления в силу настоящего постановления, действительны и подлежат замене до 1 января 2021 г. на регистрационные удостоверения нового образца.
Что касается подтверждения соответствия медицинских изделий, оно может быть в виде декларирования соответствия или обязательной сертификации. Конкретные виды медицинских изделий, которые подлежат либо декларированию либо обязательной сертификации можно найти в Постановлении Правительства РФ N 982 "Об утверждении единого перечня продукции, подлежащей обязательной сертификации, и единого перечня продукции, подтверждение соответствия которой осуществляется в форме принятия декларации о соответствии».

Обязанность проходить процедуру соответствия возложена ст. 28 Федерального закона № 184-ФЗ «О техническом регулировании».
Заявитель обязан:
– обеспечивать соответствие продукции требованиям технических регламентов;
– выпускать в обращение продукцию, подлежащую обязательному подтверждению соответствия, только после осуществления такого подтверждения соответствия;
– указывать в сопроводительной документации сведения о сертификате соответствия или декларации о соответствии;
– предъявлять в органы государственного контроля (надзора) за соблюдением требований технических регламентов, а также заинтересованным лицам документы, свидетельствующие о подтверждении соответствия продукции требованиям технических регламентов (декларацию о соответствии, сертификат соответствия или их копии) либо регистрационный номер сертификата соответствия или декларации о соответствии.
Заявитель – физическое или юридическое лицо, которое для подтверждения соответствия принимает декларацию о соответствии или обращается за получением сертификата соответствия, получает сертификат соответствия.
Информация, которая должна быть указана в товарно-сопроводительных документах, регламентирована Постановлением Правительства РФ №55 «Об утверждении Правил продажи отдельных видов товаров…»:
– о декларации (регистрационный № декларации, срок её действия, наименование организации, принявшей декларацию и орган, её зарегистрировавший);
– о сертификате (№ сертификата, срок его действия и орган, выдавший сертификат).
Посмотреть наличие действующей декларации/сертификата можно на сайте РосАккредитации в разделе «Реестры» в рубрике «Единый реестр деклараций о соответствии» либо «Единый реестр сертификатов соответствия».


Также следует обратить особое внимание на то, что копии декларации/сертификата, если поставщик не является их держателем, не несут в себе юридической силы и являются лишь информационными листами. Поэтому следует добиваться от поставщика указания в сопроводительной документации данных сведений (в официальном документе, с печатью и подписью).
Согласно ст. 37 Федерального закона №184-ФЗ «О техническом регулировании», изготовитель (исполнитель, продавец, лицо, выполняющее функции иностранного изготовителя), которому стало известно о несоответствии выпущенной в обращение продукции требованиям технических регламентов, обязан сообщить об этом в орган государственного контроля (надзора) в соответствии с его компетенцией в течение десяти дней с момента получения указанной информации.
Лицензионными требованиями и условиями, для производства медицинской техники являются:
а) соблюдение требований законодательства Российской Федерации, государственных стандартов и нормативно-технических документов по производству и контролю качества медицинской техники;
б) обязательная государственная регистрация в Российской Федерации в установленном порядке медицинской техники, заявленной соискателем для производства;
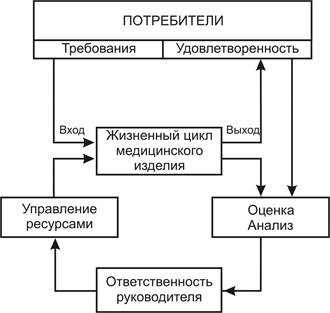
Рис.2 – Процесс управления жизненным циклом продукции
в) наличие у лицензиата принадлежащих ему на праве собственности или на ином законном основании зданий, помещений и технологического оборудования, необходимых для осуществления лицензируемой деятельности;
г) соответствие производственных помещений и оборудования техническим нормам и требованиям, предъявляемым к помещениям и оборудованию, используемым при производстве медицинской техники;
д) наличие у работников лицензиата, ответственных за производство и качество медицинской техники, высшего или среднего специального технического или медицинского образования и стажа работы по соответствующей специальности не менее 3 лет;
е) повышение не реже одного раза в 5 лет квалификации специалистов лицензиата, ответственных за производство и качество медицинской техники;
ж) наличие патентов Российской Федерации или лицензионных договоров, разрешающих производство патентованной медицинской техники.
Для получения лицензии соискатель лицензии представляет в лицензирующий орган следующие документы:
а) заявление о предоставлении лицензии;
б) копии учредительных документов и документа подтверждающего факт внесения записи о юридическом лице в Единый государственный реестр юридических лиц;
в) копия свидетельства о государственной регистрации гражданина в качестве индивидуального предпринимателя;
г) копия свидетельства о постановке соискателя лицензии на учет в налоговом органе;
д) копии документов, подтверждающих государственную регистрацию медицинской техники, которую соискатель намерен производить;
е) сведения о квалификации специалистов, ответственных за производство и качество медицинской техники;
ж) документ, подтверждающий уплату лицензионного сбора за рассмотрение заявления о предоставлении лицензии.
Дополнительные материалы, подтверждающие соблюдение лицензионных требований и условий при производстве медицинской техники:
· Номенклатура производимой медицинской техники (Приложение к заявлению)
· Копии документов, подтверждающих законность пользования производственным помещением: свидетельство о собственности, договор аренды. Договор аренды на 1 год и более должен быть зарегистрирован в соответствующем органе юстиции.
· Сведения о материально-техническом оснащении.
· Заключение санитарно-эпидемиологической службы (форма №303-00-5/у).
· Заключение противопожарной службы.
· Лицензионный договор с обладателем патента.
· Копии нормативно-технических документов по производству и контролю качества медицинской техники.
Контроль за соблюдением лицензиатом лицензионных требований и условий осуществляется в форме проверок, проводимых на основании предписания лицензирующего органа, в котором определяются лицензиат, срок проведения проверки и состав комиссии, осуществляющей проверку.
Уведомление о проведении проверки направляется лицензиату за 10 дней до ее начала.
Продолжительность проверки не должна превышать 15 дней.
Плановая проверка проводится не чаще одного раза в 2 года.
Внеплановая проверка проводится для подтверждения устранения лицензиатом выявленных при проведении плановой проверки нарушений лицензионных требований и условий, а также в случае получения лицензирующим органом информации о наличии таких нарушений.
Лицензиат обязан обеспечивать условия для проведения проверок, в том числе предоставлять необходимую информацию и документы.
По результатам проверки оформляется акт с указанием конкретных нарушений и срока их устранения.
Лицензиат обязан уведомить в письменной форме лицензирующий орган об устранении им нарушений, выявленных при проведении проверки. Проверка устранения нарушений должна быть начата не позднее 15 дней с даты получения указанного уведомления.
Производство и техобслуживание медтехники. Правовые основы
Мы уже писали о видах деятельности, подлежащих лицензированию в нашей отдельной статье. Согласно п. 17 ст. 12 закона «О лицензировании» от 04.05.2011 № 99 деятельность по производству медицинской техники и ее техобслуживанию также подлежит обязательному лицензированию. Исключением из этого правила является ситуация, в которой медтехника обслуживается юридическим лицом или индивидуальным предпринимателем исключительно для собственных нужд.
Правила лицензирования деятельности по производству и обслуживанию медтехники определены постановлением Правительства РФ «Об утверждении…» от 03.06.2013 № 469. Согласно п. 2 документа медицинская техника — это медицинские изделия (т. е. инструменты, аппараты, приборы и оборудование), применяемые в ходе осуществления медицинской деятельности отдельно или в совокупности с иными устройствами и принадлежностями (например, специализированным программным обеспечением).
Основным назначением такой техники является проведение профилактических, лечебных или реабилитационных мероприятий, выполнение исследований медицинской направленности, восстановление физиологических функций организма, их изменение или замещение, а также предотвращение или прерывание беременности. Функциональные возможности медтехники не могут быть заменены фармакологическим, генетическим, иммунологическим или иным внешним воздействием на организм человека.
Полный список существующей медицинской техники приведен в приказе Минздрава РФ «Об утверждении…» от 06.06.2012 № 4н. Производство и техобслуживание любого изделия из приведенного перечня подлежит обязательному лицензированию. Найти сведения о выданных разрешениях можно на официальном сайте ведомства, перейдя по ссылке. Для поиска необходимо ввести любые данные из указанных в поле для ввода информации (№ приказа, № входящего, ИНН, организация) либо воспользоваться полем «Расширенный поиск», где достаточно указать дату выдачи лицензии. Если найти информацию указанным выше способом не удалось, можно воспользоваться еще одним сервисом Росздравнадзора, который находится по адресу. На открывшейся странице в разделе «О предоставлении лицензий» необходимо нажать на кнопку «Смотреть все документы». Далее требуется нажать на кнопку «Расширенный поиск» и выбрать наименование органа, выдавшего лицензию, дату выдачи и интересующий раздел информации.
Лицензирование производства медицинской техники
Согласно содержанию п. 1 приложения к постановлению № 469, под производством медтехники понимается:
- непосредственно изготовление таких изделий;
- изготовление медицинской техники по заказу отдельных потребителей с целью использования ее для личных нужд конкретным пациентом.
Полномочиями по лицензированию медицинских изделий (с целью как производства, так и техобслуживания), в соответствии с п. 4 постановления № 469, обладает Федеральная служба по надзору в сфере здравоохранения (Росздравнадзор).
Процедура получения лицензии состоит из двух этапов, идущих в следующем порядке:
- Подготовка необходимой документации.
- Обращение в Росздравнадзор. Это можно сделать:
- лично обратившись в территориальное отделение ведомства;
- передав документы через представителя, предварительно оформив на его имя соответствующую доверенность;
- направив документы по почте;
- передав документы с помощью электронного сервиса (потребуется наличие личного кабинета на портале «Госуслуги»).
Принятие решения о выдаче лицензии (или отказе в этом) осуществляется в течение 45 рабочих дней с момента получения ведомством соответствующего заявления (п. 16 приказа Минздрава РФ «Об утверждении…» от 28.11.2013 № 876н). В случае положительного решения в течение 3 дней с момента внесения в реестр записи о выдаче лицензии она направляется в адрес заявителя. Размер госпошлины за выдачу лицензии составляет 7,5 тыс. руб. (подп. 92 п. 1 ст. 333.33 НК РФ).
Лицензирование технического обслуживания (ремонта) медицинской техники
Согласно п. 2 приложения к постановлению № 469, под техобслуживанием медтехники понимается:
- монтаж и наладка;
- контроль ее технического состояния;
- периодическое и текущее обслуживание;
- ремонтные работы.
Лицензирование обслуживания медицинской техники осуществляется в той же последовательности, что и лицензирование ее изготовления. Различия существуют лишь в пакете документов, представляемых в Росздравнадзор (подробная информация о его составе приведена ниже).
Заявление на получение лицензии
Чтобы осуществить лицензирование технического обслуживания медицинской техники либо ее производства, соискателю необходимо обратиться в Росздравнадзор с соответствующим заявлением. Его форма содержится в приложении 1 к приказу Росздравнадзора «Об утверждении…» от 03.04.2014 № 1271.
В заявлении необходимо указать:
- полное и сокращенное наименование юридического лица и его организационно-правовую форму или Ф. И. О. индивидуального предпринимателя и реквизиты его паспорта;
- юридический адрес (для организаций) или адрес регистрации (для ИП);
- ОГРН;
- реквизиты документа, подтверждающего факт внесения записи о заявителе в ЕГРЮЛ или ЕГРИП;
- ИНН;
- реквизиты свидетельства о постановке на налоговый учет;
- адрес места, в котором будут осуществляться работы по производству или техобслуживанию медтехники;
- вид деятельности, для осуществления которой необходима лицензия;
- реквизиты квитанции об уплате госпошлины;
- реквизиты документов, подтверждающих факт наличия у соискателя права на использование помещения для осуществления лицензируемой деятельности;
- реквизиты документов, подтверждающих наличие у соискателя права на использование средств измерений, используемых для осуществления лицензируемой деятельности;
- реквизиты документов, подтверждающих факт наличия регистрации на территории России медтехники, которую будет производить соискатель (за исключением случаев, когда такая техника производится по индивидуальному заказу);
- номер контактного телефона и адрес электронной почты;
- указание на способ выдачи лицензии (лично, по почте или через интернет).
Дополнительные документы для лицензирования
К заявлению, согласно п. 7 постановления № 469, необходимо приложить следующие документы:
- квитанцию об уплате госпошлины;
- копии документов, подтверждающих наличие у соискателя права собственности на помещения, используемые для работы с медтехникой, или иного законного основания для их использования;
- копии документов, подтверждающих факт наличия у соискателя права собственности или иного законного основания на использование средств измерений, необходимых для изготовления, ремонта или обслуживания медтехники;
- копии документов, подтверждающих факт наличия у работников соискателя, привлекаемых к производству или техобслуживанию медтехники, высшего или среднего технического образования, стажа работы по специальности не менее 3 лет, а также наличия дополнительного образования (при условии повышения квалификации не реже чем 1 раз в 5 лет)
- в случае получения лицензии на изготовление медтехники необходимо предоставить реквизиты документов, подтверждающих наличие регистрации на территории РФ тех медицинских изделий, которые соискатель планирует производить, а также копии технической и нормативной документации на медтехнику, которую будет производить заявитель;
- в случае получения лицензии на техобслуживание медтехники — эксплуатационную документацию на медтехнику, подготовленную ее производителем.
Требования, предъявляемые к соискателю лицензии
Исходя из приведенных выше сведений и положений иных нормативных актов можно сделать вывод о том, что предприятие-соискатель должно для получения лицензии обеспечить:
- Наличие в штате квалифицированных сотрудников, занятых на производстве или техобслуживании медтехники, имеющих среднее специальное или высшее образование, а также опыт работы по специальности не менее 3 лет, и проходящих курсы повышения квалификации не реже 1 раза в 5 лет.
- Наличие средств измерений, внесенных в соответствующий реестр и имеющих свидетельство о поверке и калибровке.
В том случае, если осуществляется ремонт и обслуживание стерильной медтехники, к помещениям предъявляются дополнительные требования по чистоте (с ними можно ознакомиться, изучив положения ГОСТ Р ИСО 13408-1-2000).
Последствия работы без лицензии
Выполнение работ по производству или техобслуживанию медтехники при условии отсутствия необходимой лицензии влечет за собой привлечение предпринимателя к административной или уголовной (если предприятием получен доход в крупном размере или в результате его деятельности жизни или здоровью людей был нанесен вред) ответственности.
Мера такой ответственности определяется положениями соответствующих нормативных актов:
- ч. 2 ст. 14.1 КоАП РФ — наложение штрафа в размере от 40 до 50 тыс. руб. для юр. лиц;
- ч. 1 ст. 171 УК РФ — наложение штрафа на сумму до 300 тыс. руб. или в размере заработка осужденного за период продолжительностью до 2 лет, обязательными работами длительностью до 480 часов либо арестом продолжительностью до полугода.
Итак, лицензирование медицинской техники предполагает получение разрешения на ее производство или техобслуживание (в том числе ремонт). Чтобы стать обладателем разрешительного документа, необходимо подготовить соответствующую заявку (ее форма и содержание установлены законодателем) и передать ее на рассмотрение в Росздравнадзор. В течение 45 дней с момента обращения ведомство выносит решение. В том случае, если оно будет положительным, можно приступать к выполнению работ по производству и обслуживанию медицинской продукции. Лицензирование ремонта медицинской техники или ее производства обязательно. Отсутствие лицензии влечет ответственность, предусмотренную КоАП РФ и УК РФ.
No related posts.